

Legacy Filling Lines Evolve Safeguarding with RABS Technology

A robust contamination control strategy aligned with Annex 1 (1) requirements are now crucial for aseptic manufacturing.
The utilization of restricted access barrier systems (RABS), such as open RABS, closed RABS, containment and isolator systems, plays a pivotal role in mitigating the risks and ensuring compliance with evolving regulatory standards. The updated Annex 1 provides a comprehensive elucidation of barrier technologies. Current requirements necessitate existing production lines to undertake strategic retrofitting for enhanced risk reduction and regulatory preparedness.
The filling lines in traditional aseptic filling are typically maintained as Grade A. Grade A rooms with high efficiency particulate air (HEPA) filters maintain a unidirectional airflow. Studies have established that the human factor and associated aseptic behaviors contribute to viable and nonviable contamination (2). Minimizing contamination also ensures minimal opportunities to break the "first air" (i.e., disrupt the unidirectional air stream from the HEPA-filtered air to the processing areas). Therefore, barrier technologies such as the commonly used RABS become ideal for traditional lines in minimizing unintended manipulations, minimizing the potential to break first air and, as much as possible, keeping operators away from critical processes.
Aseptic Processing: Always the Last Resort
Terminal sterilization is preferred for ensuring product sterility due to its precise sterility assurance level calculation, validation and control. Aseptic processing, however, essential for certain biologics, vaccines and injectables because of their inherent characteristics, is considered a final option. Therefore, stringent requirements are in place to minimize patient risks in aseptic manufacturing processes. A significant proportion of cleanroom contamination is attributed to human sources, with heightened activity directly impacting contamination. To mitigate this, personnel are trained, for instance, to adopt reduced movement and minimal cross-room communication practices. Recent studies have substantiated that enhanced cleanroom environment and personnel management effectively reduce particulate and microbial contamination, primarily from personnel density and activities (3). Activities conducted before entering the cleanroom can impact bioburden levels, resulting in sporadic increases in microbial counts within cleanrooms.
Water is a frequent bacterial endotoxin and bioburden source, potentially carrying gram-negative bacteria. Therefore, it is important to monitor processed water further down the chain, such as formulation or cleaning processes. Endotoxins, which originate from gram-negative bacteria, can have severe consequences when introduced into the bloodstream, leading to inflammatory responses, septic shock and fatality. These lipopolysaccharide molecules consist of a lipid, core polysaccharide and a polysaccharide side chain known as the O-antigen, which varies among bacterial strains. The lipid component anchors the endotoxin in the bacterial cell membrane, provoking inflammation during bacterial infections. Shed endotoxins bind to toll-like receptor 4 on animal cell surfaces, initiating an inflammatory response. Its heat stability necessitates ongoing monitoring of viable cells (bioburden) and endotoxins. Pharmaceutical processes and equipment face continuous risk of microbial contamination from various sources, including human contact, environmental dust, packaging materials, contaminated rinse water and microbial growth. Acceptable endotoxin levels depend on product type, disease indication, administration route and dosage quantity.
Restricted Access Barrier System
Based on the application and the status of the legacy line, an active or passive open RABS or a closed RABS (cRABS) may be suitable (4). A cRABS is preferable if controlling the room's environment and handling potent compounds are involved. An active RABS typically has the HEPA filter and fan unit all enclosed inside the barrier framework. Passive RABS, on the other hand, uses the existing room HEPA filters, making it ideal for retrofit scenarios (see Figure 1). The barrier reaches the top of the cleanroom HEPA's first-air supply in such designs. Fit-for-purpose retrofit designs provide a combination of guarding and strategic access points to keep the operator segregated and away from the first air while still enabling activities, like feeding stoppers, for processing.
Specially designed rapid transfer ports (RTPs) and other integrated transfer ports are installed to perform manipulations or to transfer materials to the interior of the RABS during aseptic operations. Doors are only opened, when necessary, after the initial setup and are monitored and documented accordingly. The inner surfaces of RABS are disinfected and decontaminated with a sporicidal agent and wiped down with 70% isopropyl alcohol (IPA) before setup operations. With newly introduced contamination controls, such as the passive RABS, reclassifying the cleanroom can also be considered from an aseptic processing Grade A cleanroom to Grade A within the barrier and maintaining a background of Grade B, provided media-fill data supports the proposed change.
 Figure 1 Active and Passive RABS
Figure 1 Active and Passive RABS

Modular-based RABS that can be customized provide better flexibility in the retrofitting process. Much attention must be given to operator ergonomics, especially from traditional to barrier-based systems, as the original space design did not consider movement within an enclosed setting. A compliant RABS should have appropriate airflow management and containment features to ensure product protection. In addition, a full risk assessment is required to identify the required features, determine additional procedures, identify routine and non-routine interventions and determine the failure modes.
When transitioning from a conventional cleanroom filling system to RABS, it is crucial to comprehensively analyze the introduction of supplementary components, such as tools, change parts, RABS gloves and mop heads (see Figure 2). They play a significant role in the functionality and operations within a RABS environment. Integrating tools into the RABS setting involves evaluating their size, shape, location and frequency of use. Tools such as tweezers, RTP tools, pushers and rakes should be strategically placed and designed to facilitate smooth operations without obstructing the barrier system or contaminating the critical environment. The selection of appropriate materials and sterilization methods for these tools is essential. New change parts for equipment adjustments or product variations are expected during such retrofitting. The change part itself should not increase contamination risks during installation or replacement. Proper handling procedures and design considerations are vital to minimize disruptions to the controlled environment. Their design, placement and utilization may impact airflow dynamics, particle dispersion and containment efficiency.
 Figure 2 Examples of RABS Components
Figure 2 Examples of RABS Components

Impact of New Interventions
During the transition from traditional filling processes to RABS, it becomes imperative to comprehensively evaluate the ramifications of the newly introduced interventions. These assessments should encompass several critical aspects, such as their effect on the validated state of the manufacturing process, its impact on smoke studies, capacity considerations, first-air concerns, the criticality of the interventions and its inclusivity in the media-fill program. One of the primary concerns is how these interventions affect the validation process for different products.
The objective is to ensure that introducing new interventions or changes does not compromise the critical setup aspects that may lead to the need to repeat the process validation and change the existing submission (e.g., the line velocity, in-process controls, etc.).
In many instances, introducing new interventions will impact existing smoke studies, requiring the repetition of these studies. New interventions can also influence the capacity of the production line. In the case of RABS systems, interventions are often expected to lead to slower processing compared to traditional lines. This slowdown can potentially affect hold times and the maximum yield of the production line, warranting close examination. Assessing whether the new designs inadvertently create concerns related to unavoidable first-air breaches is crucial. Also, interventions must be assessed in terms of the criticality of those interventions to ensure that the new interventions or the new way of doing them are not changing the registered criticality of the interventions and subsequently impacting the aseptic qualification criteria. Proper consideration must be given to the inclusivity of the media-fill program to encompass all anticipated inherent and corrective interventions. Equally important is establishing an effective process to continually update this intervention list in subsequent media-fill trials as required.
Qualification and Media Fill Strategy
A User Requirement Specification (URS) should clearly define and document the requirements of the RABS based on regulatory guidelines, manufacturer's needs and process requirements. The Design Qualification then evaluates the RABS design, including its specifications, layout, functionality and adherence to the URS. Installation Qualification verifies that the RABS has been installed correctly according to the manufacturer's design and regulatory requirements. The Operational Qualification tests confirm the system's functionality and performance under normal operating conditions to ensure it meets the defined specifications. Lastly, performance Qualification demonstrates that the RABS consistently performs according to specifications during actual operational conditions and in a simulated production environment (5).
Multiple media-fill runs with the RABS are conducted to represent different scenarios and conditions that may be encountered during actual product filling, such as aseptic interventions, equipment malfunctions and deviations that may challenge the aseptic process as prescribed by the regulatory guidelines. Under appropriate conditions, the filled media are monitored for microbial growth to determine the effectiveness of the aseptic process. The aseptic-filling process is continuously monitored, and media-fill studies need to be repeated periodically to ensure the RABS' continued effectiveness. Moreover, the challenge study requires maximum personnel to be present with an elevated activity level (as permitted by a standard operating procedure [SOP]). Case studies have attributed aseptic processing contamination to one or a combination of poor personnel practices, loss of environmental control and flawed operational design.
By following a thorough qualification and media-fill strategy, the manufacturer can demonstrate the effectiveness of the RABS in maintaining aseptic conditions during the filling process.
Cleaning Process
RABS units are cleaned manually, which involves spray and wipe-down methods for bio-decontamination. The challenge lies in achieving consistent, repeatable and complete bio-decontamination using manual methods. Now, validating the effectiveness of the cleaning and bio-decontamination solutions used in the RABS system is an important step in justifying manual cleaning processes. As part of the cleaning process validation, samples from various locations within the RABS are taken to test for cleanliness and microbial load, and glove ports for filling operations and other tasks requiring operator interaction are wiped down. The cleaning operation starts from the cleanest to the least clean areas to minimize the spread of contaminants, following the order (6):
- Ceiling
- Equipment, RABS doors inside
- Glove, RABS doors outside
- Counters, carts
- Walls, windows, doors, protruding surfaces, pass-throughs
- Floor
Appropriate personal protective equipment, including gloves, goggles, gowns and non-shedding cleaning materials and tools, are used for the cleaning operation. Special attention must be given to any movable parts and components that can come into contact with the product. The gaskets are cleaned and disinfected thoroughly, with special attention to gaskets around access points and doors. A wipe-down with 70% IPA is then followed by disinfection and contact time with a sporicidal. The routine pre-processing, post-processing, daily, weekly and monthly cleaning criteria and cleaning agent must be established. Maintaining detailed cleaning records, including dates, times, cleaning agents used, expiry date and any deviations encountered during the cleaning process, is paramount. Appropriate line clearance is performed at key stages.
Training and Documentation
The complexity of the RABS can require additional operational time. Setup activities of the RABS may include open-door and closed-door activities, requiring additional time and effort. Strict adherence to RABS protocols requires more precision and attentiveness during the filling operations, achieved through routine aseptic-practice training. Training should focus on educating personnel about the aseptic techniques required for working within the new RABS environment, including gowning, material handling and maintaining sterility. The operators need to comprehend the design, functionality and operation of the RABS. Training modules should detail the step-by-step operational procedures within the RABS, such as equipment setup, material transfer, filling processes, interventions and cleaning. In addition, it should cover contingency plans and responses to deviations or emergencies within the RABS environment. Employees should be trained in completing batch records, logbooks and any documentation specific to RABS operations.
For example, it is understood that bacterial species on the skin layer are diverse (see Figure 3), comprising more than 1,000 species (7). Further, research has established that a gowned operator has the potential to release up to 10,000 Colony Forming Units per hour (8), necessitating thorough operator training on aseptic practices (see Figure 4). A well-trained workforce contributes significantly to the success of the transition to RABS.
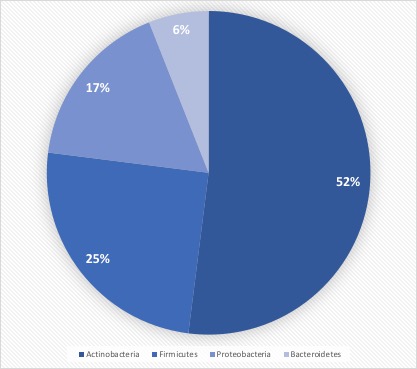 Figure 3 Human Microbiome Project
Figure 3 Human Microbiome Project

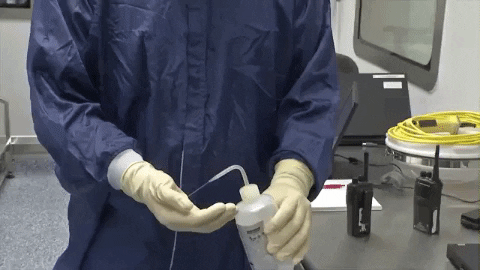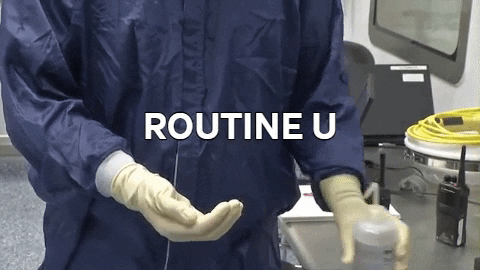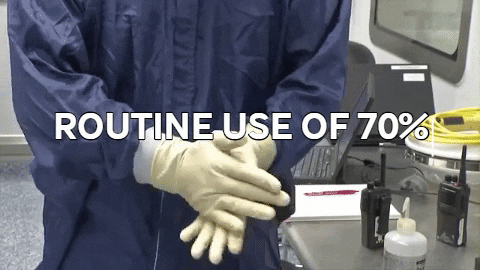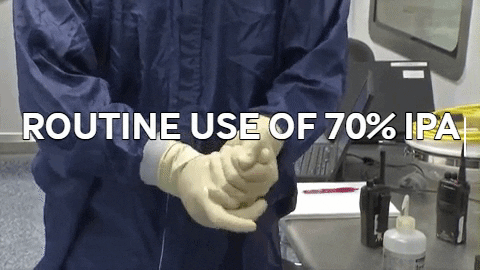 70% IPA is required to be routinely used by operators to disinfect. 70% IPA is more lethal than 100% as it slowly penetrates the cell wall and coagulates all proteins. 100% IPA evaporates fast, while 70% IPA stays active longer.
70% IPA is required to be routinely used by operators to disinfect. 70% IPA is more lethal than 100% as it slowly penetrates the cell wall and coagulates all proteins. 100% IPA evaporates fast, while 70% IPA stays active longer. Wiping in a circular motion is avoided during aseptic processing. Uni-directional wiping using parallel strokes is ideal to remove contaminants and effectivdely minimize their spread effectively. Wiping is done from the cleanest area to the least clean area.
Wiping in a circular motion is avoided during aseptic processing. Uni-directional wiping using parallel strokes is ideal to remove contaminants and effectivdely minimize their spread effectively. Wiping is done from the cleanest area to the least clean area.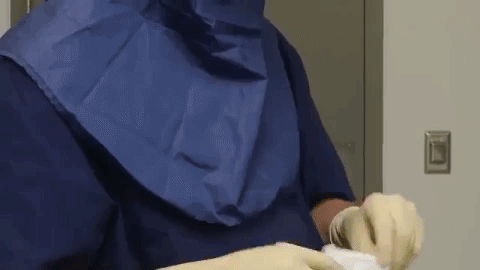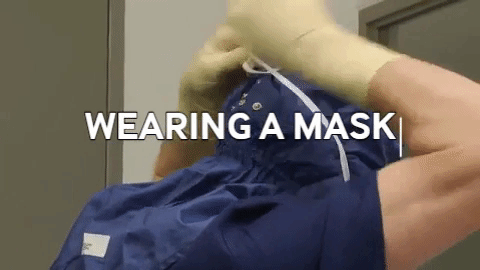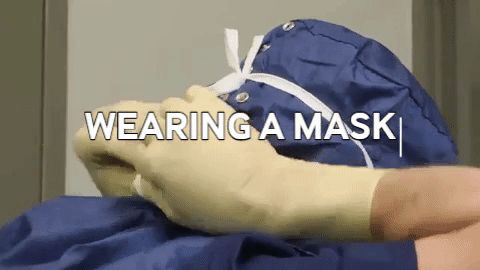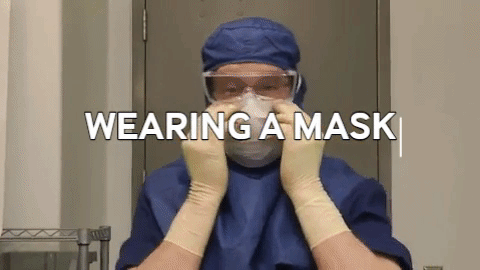 The mask is tied snugly to conform to the contour of the face. The mask is formed to the bridge of the nose. While removing and discarding, the front of the mask is not touched.
The mask is tied snugly to conform to the contour of the face. The mask is formed to the bridge of the nose. While removing and discarding, the front of the mask is not touched.When transitioning from a traditional filling process to RABS, several vital documents must be updated, revised or created to align with the new processes and technologies. New SOPs detailing the operation, maintenance, cleaning and monitoring of the RABS system are needed. Batch records must be revised, outlining the steps and procedures for filling within the RABS, including any additional checks or requirements unique to the RABS process. Records detailing the cleaning and maintenance of the RABS system, including any pre-use and post-use checks, are to be introduced. Protocols and reports are specifically designed to validate the RABS, ensuring it meets the required standards for aseptic processing. QMS documentation, such as quality policies, objectives and monitoring processes, should incorporate the changes introduced by the RABS. Environmental monitoring (EM) procedures must be re-evaluated to ensure compliance with required standards (9,10).
Regulatory Considerations
The change can impact the product’s regulatory submission status. Depending on the market, the regulatory authorities may require a thorough evaluation of the changes due to RABS implementation, potentially leading to supply disruptions. The manufacturer must work closely with regulatory authorities, clients and customers to ensure all necessary documentation is updated and submitted accurately to mitigate potential delays and associated challenges. Early information sharing about its benefits can enhance confidence in the change process. Regulatory tools such as ICH Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management (11, 12) allow for faster approval pathways by utilization of the Post-Approval Change Management Protocol (PACMP) (13). In all cases, the strategy and approach for introducing RABS to improve an aseptic filling suite for the products must be elaborated, and the acceptance criteria must be well defined.
Conclusion
Isolator technology is recognized for eliminating potential contamination from operators, leading to increased sterility assurance levels while keeping a lower Grade C background. Nevertheless, conventional cleanroom setups operating at Grade A might prefer adopting RABS technology for an upgrade, as it offers a comparable sterility assurance level with a reduced investment. They are pivotal in mitigating contamination risks and ensuring compliance with evolving regulatory standards. Minimizing contamination involves minimizing opportunities to disrupt the first air, emphasizing using barrier technologies like RABS. Active or passive RABS may be suitable based on application and the status of the legacy line, considering environmental control and handling potent compounds.
For effective product protection, retrofit designs should prioritize operator ergonomics, airflow management and containment features. Transitioning to RABS requires updating SOPs, batch records and training materials. It is crucial to involve relevant stakeholders, such as quality assurance, manufacturing, engineering and validation teams, to ensure a smooth transition to the RABS filling process. The journey from conventional aseptic practices to embracing RABS represents a technological advancement and a strategic evolution toward safer, more compliant and efficient pharmaceutical manufacturing.
References
- Volume 4 – EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Annex 1: Manufacture of Sterile Medicinal Products
- Bengt Ljungqvist and Berit ReinmÜller. People as a Contamination Source in Pharmaceutical Clean Rooms—Source Strengths and Calculated Concentrations of Airborne Contaminants, PDA J Pharm Sci Technol, Mar 2021.
- Martin S. Favero, John R. Puleo, James H. Marshall and Gordon S., Comparative Levels and Types of Microbial Contamination Detected in Industrial Clean Rooms, American Society for Microbiology, Applied Microbiology, Volume 14, Issue 4, Jul 1966.
- Advanced Aseptic Processing Technology. 2016. Switzerland: CRC Press. https://www.google.ca/books/edition/Advanced_Aseptic_Processing_Technology/rFTRBQAAQBAJ?hl=en&gbpv=0
- Ajay Babu Pazhayattil. Biopharma Equipment Design, Qualification and Monitoring Model, IVT Network, Dec 2021.
- PDA Technical Report No. 70: Fundamentals of Cleaning and Disinfection Programs for Aseptic Manufacturing Facilities, 2015.https://www.pda.org/bookstore/product-detail/2809-tr-70-cleaning-and-disinfection-programs
- NIH Human Microbiome Project Defines Normal Bacterial Makeup of the Body, News Releases, National Human Genome Research Institute, Jun 2012. https://www.nih.gov/news-events/news-releases/nih-human-microbiome-project-defines-normal-bacterial-makeup-body
- Bengt Ljungqvist and Berit ReinmÜller. People as a Contamination Source in Pharmaceutical Clean Rooms—Source Strengths and Calculated Concentrations of Airborne Contaminants, PDA Journal of Pharmaceutical Science and Technology March 2021, 75(2), 119-127
- PDA Technical Report No. 13 (Revised 2022): Fundamentals of an Environmental Monitoring Program, 2022.
https://www.pda.org/bookstore/product-detail/6644-tr-13-revised-2022-fund-environmental-monitoring
BioPhorum. Environmental Monitoring (EM): A Harmonized Risk-Based Approach to Selecting Monitoring Points and Defining Monitoring Plans, Nov 2020.https://www.biophorum.com/download/environmental-monitoring-em-a-harmonized-risk-based-approach-to-selecting-monitoring-points-and-defining-monitoring-plans/
ICH Quality Guideline Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management https://database.ich.org/sites/default/files/Q12_Guideline_Step4_2019_1119.pdf
12. PDA Technical Report No. 91: Post-Approval Change Management and Implementation for Biologics and Pharmaceutical Drug Products — A User’s Guide, 2023. https://www.pda.org/bookstore/product-detail/7396-tr-91-post-approval-change-management
Naheed Sayeed-Desta, Ajay Babu Pazhayattil, and Ivy Louis. Prospects for Post-Approval Change Management, PDA Letter, May 29, 2018. https://www.pda.org/pda-letter-portal/home/full-article/prospects-for-post-approval-change-management
About the Authors
-

Ahmed Elsaid, Emergent Biosolution
Elsaid is a Quality Senior Director at Emergent Biosolution in Rockville, Maryland. Elsaid has over 22 years of experience in the pharmaceutical industry as a quality leader in parenteral manufacturing. He has diverse experience working on four different continents addressing the different regulations and managing projects in different phases of product lifecycles. Elsaid’s experience spans multiple disciplines, including quality systems, aseptic processing, validation, analytical and process development, commissioning and qualifications.
-

Ajay Pazhayattil, PhD, cGMP World
Pazhayattil provides strategic direction and leadership to brand, generic, biosimilar and CDMO organizations. Pazhayattil is an industrial pharmacist and a subject matter expert who has provided tangible results across pharmaceutical and biopharmaceutical technical operations, validation, quality, regulatory and operations segments. Pazhayattil is an author, speaker, committee lead and influencer in the industry.